Usage¶
The SNP Pipeline is run from the Unix command line. The pipeline consists of multiple commands.
| Command | Description |
|---|---|
| run_snp_pipeline.sh | This do-it-all script runs the other scripts listed below, comprising all the pipeline steps. This is the same as running cfsan_snp_pipeline run. |
| cfsan_snp_pipeline | This is the main command line tool with subcommands listed below |
| run | This all-in-one command runs all the necessary pipeline subcommands listed below, in the right order, comprising all the pipeline steps |
| data | Copies supplied example data to a work directory |
| index_ref | Indexes the reference genome |
| map_reads | Aligns sample reads to the reference genome |
| call_sites | Finds high-confidence SNPs in each sample |
| filter_regions | Remove SNPs in abnormal regions |
| merge_sites | Combines the SNP positions across all samples into a single unified SNP list file |
| call_consensus | Calls the consensus SNPs for each sample |
| merge_vcfs | Creates a multi-sample VCF file with the SNPs found in all samples |
| snp_matrix | Creates a matrix of SNPs across all samples |
| distance | Computes the SNP distances between all pairs of samples |
| snp_reference | Writes the reference sequence bases at SNP locations to a fasta file |
| collect_metrics | Collects useful coverage and variant statistics about each sample |
| combine_metrics | Creates a table of coverage and variant statistics for all samples |
| purge | Removes the intermediate output files in the “samples” directory to save space upon successful completion of a SNP Pipeline run |
Inputs¶
Before using the SNP Pipeline, make sure your input data is organized and named the way the pipeline expects. Follow these guidelines:
No spaces in file names and directory names.
A fasta genome reference file must exist in a separate directory.
The samples must be organized with a separate directory for each sample. Each sample directory should contain the fastq files for that sample. The name of the directory should match the name of the sample. When using paired-end fastq files, the forward and reverse files must be in the same directory.
The script needs to know how to find all the samples. You have two choices:
- You can organize all the sample directories under a common parent directory.
- You can have sample directories anywhere you like, but you will need to create a file listing the path to all the sample directories.
The sample fastq files must be named with one of the following file patterns: (*.fastq, *.fq, *.fastq.gz, *.fq.gz). It’s okay if different samples are named differently, but the two mate files of paired-end samples must be named with the same extension.
If there is an outgroup among samples, a file containing the sample ids of the outgroup samples must be created in advance, and the relative or absolute path of this file should be specified in the parameter “FilterRegions_ExtraParams” in the configuration file “snppipeline.conf” (see the description of the parameter “FilterRegions_ExtraParams”).
Outputs¶
By default, the SNP Pipeline generates the following output files. If you need more control over the output, you can run the pipeline one step at a time (not recommended). See Step-by-Step Workflows.
snplist.txt: contains a list of the high-confidence SNP positions identified by the phase 1 SNP caller (VarScan) in at least one of the samples. These are the only positions where the consensus caller subsequently looks for SNPs in all samples. The consensus caller often finds SNPs at the same positions in other samples, and those additional SNPs are not listed in the snplist.txt file. While the snplist.txt file has an accurate list of SNP positions, it does not contain the final list of samples having SNPs at those positions. If you need the final set of SNPs per sample, you should not use the snplist.txt file. Instead, refer to the snpma.fasta file or the snpma.vcf file. The correspondingsnplist_preserved.txtfile is produced when snp filtering removes the abnormal snps.consensus.fasta: for each sample, the consensus base calls at the high-confidence positions where SNPs were detected in any of the samples. The correspondingconsensus_preserved.fastafile is produced when snp filtering removes the abnormal snps.consensus.vcf: for each sample, the VCF file of SNPs called, as well as failed SNPs at the high-confidence positions where SNPs were detected in any of the samples. The correspondingconsensus_preserved.vcffile is produced when snp filtering removes the abnormal snps.snpma.fasta: the SNP matrix containing the consensus base for each of the samples at the high-confidence positions where SNPs were identified in any of the samples. The matrix contains one row per sample and one column per SNP position. Non-SNP positions are not included in the matrix. The matrix is formatted as a fasta file, with each sequence (all of identical length) corresponding to the SNPs in the correspondingly named sequence. The correspondingsnpma_preserved.fastafile is produced when snp filtering removes the abnormal snps.snp_distance_pairwise.tsv: contains the pairwise SNP distance between all pairs of samples. The file is tab-separated, with a header row and three columns identifing the two sequences and their distance. The correspondingsnp_distance_pairwise_preserved.tsvfile is produced when snp filtering removes the abnormal snps.snp_distance_matrix.tsv: contains a matrix of the SNP distances between all pairs of samples. The file is tab-separated, with a header row and rows and columns for all samples. The correspondingsnp_distance_matrix_preserved.tsvfile is produced when snp filtering removes the abnormal snps.snpma.vcf: contains the merged multi-sample VCF file identifying the positions and snps for all samples. The correspondingsnpma_preserved.vcffile is produced when snp filtering removes the abnormal snps.referenceSNP.fasta: a fasta file containing the reference sequence bases at all the SNP locations. The correspondingreferenceSNP_preserved.fastafile is produced when snp filtering removes the abnormal snps.metrics: for each sample, contains the size of the sample, number of reads, alignment rate, pileup depth, and number of SNPs found.metrics.tsv: a tab-separated table of metrics for all samples containing the size of the samples, number of reads, alignment rate, pileup depth, and number of SNPs found.error.log: a summary of errors detected during SNP Pipeline execution
All-In-One SNP Pipeline Script¶
Most users should run the SNP Pipeline by executing the cfsan_snp_pipeline run command.
This command is easy to use and works equally well on
your desktop workstation or on a High Performance Computing cluster. You can
find examples of using the command in the sections below.
If you need more flexibility, you can run the individual pipeline scripts one step at a time (not recommended). See Step-by-Step Workflows.
Logging¶
The SNP Pipeline generates log files for each processing step of the pipeline. The logs for each pipeline run are stored in a time-stamped directory under the output directory. If the pipeline is re-run on the same samples, the old log files are kept and a new log directory is created for the new run. For example, the output directory might look like this after two runs:
drwx------ 2 me group 4096 Oct 17 16:37 logs-20141017.154428/
drwx------ 2 me group 4096 Oct 17 16:38 logs-20141017.163848/
drwx------ 2 me group 4096 Oct 17 16:37 reference/
-rw------- 1 me group 194 Oct 17 16:38 referenceSNP.fasta
-rw------- 1 me group 182 Oct 17 16:38 referenceSNP_preserved.fasta
-rw------- 1 me group 104 Oct 17 16:38 sampleDirectories.txt
drwx------ 6 me group 4096 Oct 17 16:37 samples/
-rw------- 1 me group 7216 Oct 17 16:38 snplist.txt
-rw------- 1 me group 6824 Oct 17 16:38 snplist_preserved.txt
-rw------- 1 me group 708 Oct 17 16:38 snpma.fasta
-rw------- 1 me group 682 Oct 17 16:38 snpma_preserved.fasta
A log file is created for each step of the pipeline for each sample. For performance reasons, the samples are sorted by size and processed largest first. This sorting is reflected in the naming of the log files. The log files are named with a suffix indicating the sample number:
-rw------- 1 me group 12045 Oct 17 16:37 indexRef.log
-rw------- 1 me group 1330 Oct 17 16:37 mapReads.log-1
-rw------- 1 me group 1330 Oct 17 16:37 mapReads.log-2
-rw------- 1 me group 1330 Oct 17 16:37 mapReads.log-3
-rw------- 1 me group 1686 Oct 17 16:37 callSites.log-1
-rw------- 1 me group 1686 Oct 17 16:37 callSites.log-2
-rw------- 1 me group 1686 Oct 17 16:37 callSites.log-3
-rw------- 1 me group 1035 Oct 17 16:37 filterRegions.log
-rw------- 1 me group 1275 Oct 17 16:37 mergeSites.log
-rw------- 1 me group 1375 Oct 17 16:37 mergeSites_preserved.log
-rw------- 1 me group 1509 Oct 17 16:37 callConsensus.log-1
-rw------- 1 me group 1509 Oct 17 16:37 callConsensus.log-2
-rw------- 1 me group 1509 Oct 17 16:37 callConsensus.log-3
-rw------- 1 me group 1509 Oct 17 16:37 callConsensus_preserved.log-1
-rw------- 1 me group 1509 Oct 17 16:37 callConsensus_preserved.log-2
-rw------- 1 me group 1509 Oct 17 16:37 callConsensus_preserved.log-3
-rw------- 1 me group 1047 Oct 17 16:37 snpMatrix.log
-rw------- 1 me group 1147 Oct 17 16:37 snpMatrix_preserved.log
-rw------- 1 me group 806 Oct 17 16:37 snpReference.log
-rw------- 1 me group 806 Oct 17 16:37 snpReference_preserved.log
-rw------- 1 me group 1895 Oct 17 16:37 mergeVcfs.log
-rw------- 1 me group 2039 Oct 17 16:37 mergeVcfs_preserved.log
-rw------- 1 me group 887 Oct 17 16:37 distance.log
-rw------- 1 me group 977 Oct 17 16:37 distance_preserved.log
-rw------- 1 me group 2169 Oct 17 16:37 collectMetrics.log-1
-rw------- 1 me group 2169 Oct 17 16:37 collectMetrics.log-2
-rw------- 1 me group 2169 Oct 17 16:37 collectMetrics.log-3
-rw------- 1 me group 983 Oct 17 16:37 combineMetrics.log
To determine which samples correspond to which log files, you can either grep the log files for the sample name or inspect the sorted sampleDirectories.txt file to determine the sequential position of the sample. The file names are consistent regardless of whether the pipeline is run on a workstation or HPC cluster.
In addition to the processing log files, the log directory also contains a copy of the configuration file used for each run – capturing the parameters used during the run.
Mirrored Inputs¶
The SNP Pipeline has the optional capability to create a mirrored copy of the input fasta and fastq files. You might use this feature to avoid polluting the reference directory and sample directories with the intermediate files generated by the snp pipeline. The mirroring function can either create normal copies of the files, or it can create links to the original files – saving both time and disk space. With linked files, you can easily run multiple experiments on the same data or different overlapping sets of samples without having duplicate copies of the original sample files. See the run command reference for the mirroring syntax.
The mirroring function creates a “reference” subdirectory and a “samples” subdirectory under the main output directory. One directory per sample is created under the “samples” directory. The generated intermediate files are placed into the mirrored directories, not in the original locations of the inputs. The SNP Pipeline attempts to preserve the time stamps of the original files in the mirrored directories.
Keep in mind the following limitations when mirroring the inputs.
- Some file systems do not support soft (symbolic) links. If you attempt to create a soft link on a file system without the capability, the operation will fail with an error message.
- Hard links cannot be used to link files across two different file systems. The original file and the link must both reside on the same file system.
- Normal file copies should always work, but the copy operation can be lengthy and the duplicate files will consume extra storage space.
High Performance Computing¶
The SNP Pipeline can be executed on a High Performance Computing cluster. The Grid Engine, SLURM, and Torque job queue managers are supported.
Grid Engine¶
To run the SNP Pipeline on grid engine you must use a configuration file to specify the name of your parallel environment.
Grab the default configuration file:
cfsan_snp_pipeline data configurationFile
Edit the snppipeline.conf file and make the following change:
GridEngine_PEname="myPE" # substitute the name of your PE
You may also need to change the GridEngine_StripJobArraySuffix configuration parameter if
you see qsub illegal dependency errors.
Then run the pipeline with the -c and -Q command line options:
cfsan_snp_pipeline run -c snppipeline.conf -Q grid -s mySamplesDir myReference.fasta
You can pass extra options to the Grid Engine qsub command by configuring the GridEngine_QsubExtraParams
parameter in the configuration file. Among other things, you can control which queue the
snp-pipeline will use when executing on an HPC with multiple queues.
SLURM¶
To run the SNP Pipeline on SLURM:
cfsan_snp_pipeline run -Q slurm -s mySamplesDir myReference.fasta
You can pass extra options to the SLURM sbatch command by configuring the Slurm_SbatchExtraParams
parameter in the configuration file.
Torque¶
To run the SNP Pipeline on torque:
cfsan_snp_pipeline run -Q torque -s mySamplesDir myReference.fasta
You may need to change the Torque_StripJobArraySuffix configuration parameter if
you see qsub illegal dependency errors.
You can pass extra options to the Torque qsub command by configuring the Torque_QsubExtraParams
parameter in the configuration file.
See also: Performance.
Tool Selection¶
The SNP Pipeline lets you choose either the Bowtie2 aligner or the Smalt aligner. Your choice of aligner, as well as the command line options for the aligner are specified in the SNP Pipeline configuration file.
Grab the default configuration file:
cfsan_snp_pipeline data configurationFile
To run the SNP Pipeline with Bowtie2, edit snppipeline.conf with these settings:
SnpPipeline_Aligner="bowtie2"
Bowtie2Build_ExtraParams="" # substitute the command line options you want here
Bowtie2Align_ExtraParams="" # substitute the command line options you want here
To run the SNP Pipeline with Smalt, edit snppipeline.conf with these settings:
SnpPipeline_Aligner="smalt"
SmaltIndex_ExtraParams="" # substitute the command line options you want here
SmaltAlign_ExtraParams="" # substitute the command line options you want here
Then run the pipeline with the -c command line option:
cfsan_snp_pipeline run -c snppipeline.conf -s mySamplesDir myReference.fasta
See also Configuration.
All-In-One SNP Pipeline Workflows¶
The sections below give detailed examples of workflows you can execute with the
all-in-one run command.
All-In-One Workflow - Lambda Virus¶
The SNP Pipeline software distribution includes a small Lambda Virus data set that can be quickly processed to verify the basic functionality of the software.
Step 1 - Gather data:
# The SNP Pipeline distribution includes sample data organized as shown below:
snppipeline/data/lambdaVirusInputs/reference/lambda_virus.fasta
snppipeline/data/lambdaVirusInputs/samples/sample1/sample1_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample1/sample1_2.fastq
snppipeline/data/lambdaVirusInputs/samples/sample2/sample2_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample2/sample2_2.fastq
snppipeline/data/lambdaVirusInputs/samples/sample3/sample3_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample3/sample3_2.fastq
snppipeline/data/lambdaVirusInputs/samples/sample4/sample4_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample4/sample4_2.fastq
# Copy the supplied test data to a work area:
cd test
cfsan_snp_pipeline data lambdaVirusInputs testLambdaVirus
cd testLambdaVirus
Step 2 - Run the SNP Pipeline:
# Run the pipeline, specifing the locations of samples and the reference
#
# Specify the following options:
# -s : samples parent directory
cfsan_snp_pipeline run -s samples reference/lambda_virus.fasta
Step 3 - View and verify the results:
Upon successful completion of the pipeline, the snplist.txt file should have 166 entries, and the snplist_preserved.txt should have 137 entries. The SNP Matrix can be found in snpma.fasta and snpma_preserved.fasta. The corresponding reference bases are in the referenceSNP.fasta and referenceSNP_preserved.fasta:
# Verify the result files were created
ls -l snplist.txt
ls -l snpma.fasta
ls -l snpma.vcf
ls -l referenceSNP.fasta
ls -l snp_distance_matrix.tsv
ls -l snplist_preserved.txt
ls -l snpma_preserved.fasta
ls -l snpma_preserved.vcf
ls -l referenceSNP_preserved.fasta
ls -l snp_distance_matrix_preserved.tsv
# Verify correct results
cfsan_snp_pipeline data lambdaVirusExpectedResults expectedResults
diff -q -s snplist.txt expectedResults/snplist.txt
diff -q -s snpma.fasta expectedResults/snpma.fasta
diff -q -s referenceSNP.fasta expectedResults/referenceSNP.fasta
diff -q -s snp_distance_matrix.tsv expectedResults/snp_distance_matrix.tsv
diff -q -s snplist_preserved.txt expectedResults/snplist_preserved.txt
diff -q -s snpma_preserved.fasta expectedResults/snpma_preserved.fasta
diff -q -s referenceSNP_preserved.fasta expectedResults/referenceSNP_preserved.fasta
diff -q -s snp_distance_matrix_preserved.tsv expectedResults/snp_distance_matrix_preserved.tsv
# View the per-sample metrics
xdg-open metrics.tsv
All-In-One Workflow - Salmonella Agona¶
The Salmonella Agona data set contains a small number of realistic sequences that can be processed in a reasonable amount of time. Due to the large size of real data, the sequences must be downloaded from the NCBI SRA. Follow the instructions below to download and process the data set.
Step 1 - Gather data:
# The SNP Pipeline distribution does not include the sample data, but does
# include information about the sample data, as well as the reference
# sequence. The files are organized as shown below:
snppipeline/data/agonaInputs/sha256sumCheck
snppipeline/data/agonaInputs/reference/NC_011149.fasta
snppipeline/data/agonaInputs/sampleList
# Copy the supplied test data to a work area:
mkdir testAgona
cd testAgona
cfsan_snp_pipeline data agonaInputs cleanInputs
cd cleanInputs
# Create sample directories and download sample data from SRA at NCBI. Note that
# we use the fastq-dump command from the NCBI SRA-toolkit to fetch sample
# sequences. There are other ways to get the data, but the SRA-toolkit is
# easy to install, and does a good job of downloading large files.
mkdir samples
< sampleList xargs -I % sh -c 'mkdir samples/%; fastq-dump --gzip --origfmt --split-files --outdir samples/% %;'
# Check the data
# The original data was used to generate a hash as follows:
# sha256sum sampleList reference/*.fasta samples/*/*.fastq.gz > sha256sumCheck
# The command below checks the downloaded data (and the reference sequence) against the
# hashes that are saved in the sha256sumCheck file using sha256sum command, which is
# generally available on unix systems.
sha256sum -c sha256sumCheck
cd ..
Step 2 - Run the SNP Pipeline:
# Run the pipeline
# Specify the following options:
# -m : mirror the input samples and reference files
# -o : output directory
# -s : samples parent directory
cfsan_snp_pipeline run -m soft -o outputDirectory -s cleanInputs/samples cleanInputs/reference/NC_011149.fasta
Step 3 - View and verify the results:
Upon successful completion of the pipeline, the snplist.txt file should have 2620 entries, and the snplist_preserved.txt should have 233 entries. The SNP Matrix can be found in snpma.fasta. The corresponding reference bases are in the files referenceSNP.fasta and referenceSNP_preserved.fasta:
# Verify the result files were created
ls -l outputDirectory/snplist.txt
ls -l outputDirectory/snpma.fasta
ls -l outputDirectory/snpma.vcf
ls -l outputDirectory/referenceSNP.fasta
ls -l outputDirectory/snp_distance_matrix.tsv
ls -l outputDirectory/snplist_preserved.txt
ls -l outputDirectory/snpma_preserved.fasta
ls -l outputDirectory/snpma_preserved.vcf
ls -l outputDirectory/referenceSNP_preserved.fasta
ls -l outputDirectory/snp_distance_matrix_preserved.tsv
# Verify correct results
cfsan_snp_pipeline data agonaExpectedResults expectedResults
diff -q -s outputDirectory/snplist.txt expectedResults/snplist.txt
diff -q -s outputDirectory/snpma.fasta expectedResults/snpma.fasta
diff -q -s outputDirectory/referenceSNP.fasta expectedResults/referenceSNP.fasta
diff -q -s outputDirectory/snp_distance_matrix.tsv expectedResults/snp_distance_matrix.tsv
diff -q -s outputDirectory/snplist_preserved.txt expectedResults/snplist_preserved.txt
diff -q -s outputDirectory/snpma_preserved.fasta expectedResults/snpma_preserved.fasta
diff -q -s outputDirectory/referenceSNP_preserved.fasta expectedResults/referenceSNP_preserved.fasta
diff -q -s outputDirectory/snp_distance_matrix_preserved.tsv expectedResults/snp_distance_matrix_preserved.tsv
# View the per-sample metrics
xdg-open outputDirectory/metrics.tsv
All-In-One Workflow - Listeria monocytogenes¶
This Listeria monocytogene data set is based on an oubreak investigation related to contamination in stone fruit. It only contains environmental/produce isolates, though the full investigation contained data obtained from clinical samples as well. Due to the large size of the data, the sequences must be downloaded from the NCBI SRA. The instructions below show how to create the data set and process it.
This workflow illustrates how to run the SNP Pipeline on a High Performance Computing
cluster (HPC) running the Torque job queue manager. If you do not have a cluster available,
you can still work through this example – just remove the -Q torque command line
option in step 2.
Step 1 - Create dataset:
# The SNP Pipeline distribution does not include the sample data, but does
# include information about the sample data, as well as the reference
# sequence. The files are organized as shown below:
snppipeline/data/listeriaInputs/sha256sumCheck
snppipeline/data/listeriaInputs/reference/CFSAN023463.HGAP.draft.fasta
snppipeline/data/listeriaInputs/sampleList
# Copy the supplied test data to a work area:
mkdir testDir
cd testDir
cfsan_snp_pipeline data listeriaInputs cleanInputs
cd cleanInputs
# Create sample directories and download sample data from SRA at NCBI. Note that
# we use the fastq-dump command from the NCBI SRA-toolkit to fetch sample
# sequences. There are other ways to get the data, but the SRA-toolkit is
# easy to install, and does a good job of downloading large files.
mkdir samples
< sampleList xargs -I % sh -c ' mkdir samples/%; fastq-dump --gzip --split-files --outdir samples/% %;'
# Check the data
# The original data was used to generate a hash as follows:
# sha256sum sampleList reference/*.fasta samples/*/*.fastq.gz > sha256sumCheck
# The command below checks the downloaded data (and the reference sequence) against the
# hashes that are saved in the sha256sumCheck file using sha256sum command, which is
# generally available on unix systems.
sha256sum -c sha256sumCheck
cd ..
Step 2 - Run the SNP Pipeline:
There are a couple of parameters you may need to adjust for this analysis or other analysis
work that your do. These parameters are the number of CPU cores that are used, and the
amount of memory that is used by the java virtual machine. Both can be set in a
configuration file you can pass to cfsan_snp_pipeline run with the -c option.
See Performance.
Launch the pipeline:
# Run the pipeline.
# Specify the following options:
# -m : mirror the input samples and reference files
# -Q : HPC job queue manager
# -o : output directory
# -s : samples parent directory
cfsan_snp_pipeline run -m soft -Q torque -o outputDirectory -s cleanInputs/samples cleanInputs/reference/CFSAN023463.HGAP.draft.fasta
Step 3 - View and verify the results:
Upon successful completion of the pipeline, the snplist.txt file should have 10,102 entries, and the snplist_preserved.txt file should have 1,040 entries. The SNP Matrix can be found in snpma.fasta and snpma_preserved.fasta. The corresponding reference bases are in the referenceSNP.fasta and referenceSNP_preserved.fasta:
# Verify the result files were created
ls -l outputDirectory/snplist.txt
ls -l outputDirectory/snpma.fasta
ls -l outputDirectory/snpma.vcf
ls -l outputDirectory/referenceSNP.fasta
ls -l outputDirectory/snp_distance_matrix.tsv
ls -l outputDirectory/snplist_preserved.txt
ls -l outputDirectory/snpma_preserved.fasta
ls -l outputDirectory/snpma_preserved.vcf
ls -l outputDirectory/referenceSNP_preserved.fasta
ls -l outputDirectory/snp_distance_matrix_preserved.tsv
# Verify correct results
cfsan_snp_pipeline data listeriaExpectedResults expectedResults
diff -q -s outputDirectory/snplist.txt expectedResults/snplist.txt
diff -q -s outputDirectory/snpma.fasta expectedResults/snpma.fasta
diff -q -s outputDirectory/referenceSNP.fasta expectedResults/referenceSNP.fasta
diff -q -s outputDirectory/snp_distance_matrix.tsv expectedResults/snp_distance_matrix.tsv
diff -q -s outputDirectory/snplist_preserved.txt expectedResults/snplist_preserved.txt
diff -q -s outputDirectory/snpma_preserved.fasta expectedResults/snpma_preserved.fasta
diff -q -s outputDirectory/referenceSNP_preserved.fasta expectedResults/referenceSNP_preserved.fasta
diff -q -s outputDirectory/snp_distance_matrix_preserved.tsv expectedResults/snp_distance_matrix_preserved.tsv
# View the per-sample metrics
xdg-open outputDirectory/metrics.tsv
Step-by-Step Workflows¶
The cfsan_snp_pipeline run command described above provides a simple and powerful interface
for running all the pipeline steps from a single command. If you need more
control over the inputs, outputs, or processing steps, you can run the pipeline
one step at a time, however this is not recommended.
The sections below give detailed examples of workflows you can run with the component tools of the pipeline.
Step-by-Step Workflow - Lambda Virus¶
The SNP Pipeline software distribution includes a small Lambda Virus data set that can be quickly processed to verify the basic functionality of the software.
Step 1 - Gather data:
# The SNP Pipeline distribution includes sample data organized as shown below:
snppipeline/data/lambdaVirusInputs/reference/lambda_virus.fasta
snppipeline/data/lambdaVirusInputs/samples/sample1/sample1_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample1/sample1_2.fastq
snppipeline/data/lambdaVirusInputs/samples/sample2/sample2_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample2/sample2_2.fastq
snppipeline/data/lambdaVirusInputs/samples/sample3/sample3_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample3/sample3_2.fastq
snppipeline/data/lambdaVirusInputs/samples/sample4/sample4_1.fastq
snppipeline/data/lambdaVirusInputs/samples/sample4/sample4_2.fastq
# Copy the supplied test data to a work area:
cd test
cfsan_snp_pipeline data lambdaVirusInputs testLambdaVirus
cd testLambdaVirus
Step 2 - Prep work:
# Create files of sample directories and fastQ files:
ls -d samples/* > sampleDirectories.txt
rm sampleFullPathNames.txt 2>/dev/null
cat sampleDirectories.txt | while read dir; do echo $dir/*.fastq* >> sampleFullPathNames.txt; done
# Determine the number of CPU cores in your computer
numCores=$(grep -c ^processor /proc/cpuinfo 2>/dev/null || sysctl -n hw.ncpu)
Step 3 - Prep the reference:
cfsan_snp_pipeline index_ref reference/lambda_virus.fasta
Step 4 - Align the samples to the reference:
# Align each sample, one at a time, using all CPU cores
export Bowtie2Align_ExtraParams="--reorder -X 1000"
export SamtoolsSamFilter_ExtraParams="-F 4 -q 30"
cat sampleFullPathNames.txt | xargs -n 2 -L 1 cfsan_snp_pipeline map_reads --threads $numCores reference/lambda_virus.fasta
Step 5 - Find the sites having high-confidence SNPs:
# Process the samples in parallel using all CPU cores
export SamtoolsMpileup_ExtraParams="-q 0 -Q 13 -A"
export VarscanMpileup2snp_ExtraParams="--min-var-freq 0.90"
cat sampleDirectories.txt | xargs -n 1 -P $numCores cfsan_snp_pipeline call_sites reference/lambda_virus.fasta
Step 6 - Identify regions with abnormal SNP density and remove SNPs in these regions:
cfsan_snp_pipeline filter_regions --window_size 1000 125 15 --max_snp 3 2 1 -n var.flt.vcf sampleDirectories.txt reference/lambda_virus.fasta
Step 7 - Combine the SNP positions across all samples into the SNP list file:
cfsan_snp_pipeline merge_sites -n var.flt.vcf -o snplist.txt sampleDirectories.txt sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline merge_sites -n var.flt_preserved.vcf -o snplist_preserved.txt sampleDirectories.txt sampleDirectories.txt.PresVCF.filtered
Step 8 - Call the consensus base at SNP positions for each sample:
# Process the samples in parallel using all CPU cores
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline call_consensus -l snplist.txt --minConsDpth 3 --vcfFileName consensus.vcf --vcfRefName lambda_virus.fasta -o XX/consensus.fasta XX/reads.all.pileup
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline call_consensus -l snplist_preserved.txt --minConsDpth 3 --vcfFileName consensus_preserved.vcf --vcfRefName lambda_virus.fasta -o XX/consensus_preserved.fasta -e XX/var.flt_removed.vcf XX/reads.all.pileup
Step 9 - Create the SNP matrix:
cfsan_snp_pipeline snp_matrix -c consensus.fasta -o snpma.fasta sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline snp_matrix -c consensus_preserved.fasta -o snpma_preserved.fasta sampleDirectories.txt.PresVCF.filtered
Step 10 - Create the reference base sequence:
cfsan_snp_pipeline snp_reference -l snplist.txt -o referenceSNP.fasta reference/lambda_virus.fasta
cfsan_snp_pipeline snp_reference -l snplist_preserved.txt -o referenceSNP_preserved.fasta reference/lambda_virus.fasta
Step 11 - Collect metrics for each sample:
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline collect_metrics -o XX/metrics XX reference/lambda_virus.fasta
Step 12 - Tabulate the metrics for all samples:
cfsan_snp_pipeline combine_metrics -n metrics -o metrics.tsv sampleDirectories.txt
Step 13 - Merge the VCF files for all samples into a multi-sample VCF file:
cfsan_snp_pipeline merge_vcfs -n consensus.vcf -o snpma.vcf sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline merge_vcfs -n consensus_preserved.vcf -o snpma_preserved.vcf sampleDirectories.txt.PresVCF.filtered
Step 14 - Compute the SNP distances between samples:
cfsan_snp_pipeline distance -p snp_distance_pairwise.tsv -m snp_distance_matrix.tsv snpma.fasta
cfsan_snp_pipeline distance -p snp_distance_pairwise_preserved.tsv -m snp_distance_matrix_preserved.tsv snpma_preserved.fasta
Step 15 - View and verify the results:
Upon successful completion of the pipeline, the snplist.txt file should have 166 entries. The SNP Matrix can be found in snpma.fasta. The corresponding reference bases are in the referenceSNP.fasta file:
# Verify the result files were created
ls -l snplist.txt
ls -l snpma.fasta
ls -l snpma.vcf
ls -l referenceSNP.fasta
ls -l snp_distance_matrix.tsv
ls -l snplist_preserved.txt
ls -l snpma_preserved.fasta
ls -l snpma_preserved.vcf
ls -l referenceSNP_preserved.fasta
ls -l snp_distance_matrix_preserved.tsv
# Verify correct results
cfsan_snp_pipeline data lambdaVirusExpectedResults expectedResults
diff -q -s snplist.txt expectedResults/snplist.txt
diff -q -s snpma.fasta expectedResults/snpma.fasta
diff -q -s referenceSNP.fasta expectedResults/referenceSNP.fasta
diff -q -s snp_distance_matrix.tsv expectedResults/snp_distance_matrix.tsv
diff -q -s snplist_preserved.txt expectedResults/snplist_preserved.txt
diff -q -s snpma_preserved.fasta expectedResults/snpma_preserved.fasta
diff -q -s referenceSNP_preserved.fasta expectedResults/referenceSNP_preserved.fasta
diff -q -s snp_distance_matrix_preserved.tsv expectedResults/snp_distance_matrix_preserved.tsv
# View the per-sample metrics
xdg-open metrics.tsv
Step-by-Step Workflow - Salmonella Agona¶
The Salmonella Agona data set contains realistic sequences that can be processed in a reasonable amount of time. Due to the large size of real data, the sequences must be downloaded from the NCBI SRA. Follow the instructions below to download and process the data set.
Step 1 - Gather data:
# The SNP Pipeline distribution does not include the sample data, but does
# include information about the sample data, as well as the reference
# sequence. The files are organized as shown below:
snppipeline/data/agonaInputs/sha256sumCheck
snppipeline/data/agonaInputs/reference/NC_011149.fasta
snppipeline/data/agonaInputs/sampleList
# Copy the supplied test data to a work area:
mkdir testAgona
cd testAgona
cfsan_snp_pipeline data agonaInputs .
# Create sample directories and download sample data from SRA at NCBI. Note that
# we use the fastq-dump command from the NCBI SRA-toolkit to fetch sample
# sequences. There are other ways to get the data, but the SRA-toolkit is
# easy to install, and does a good job of downloading large files.
mkdir samples
< sampleList xargs -I % sh -c 'mkdir samples/%; fastq-dump --gzip --origfmt --split-files --outdir samples/% %;'
# Check the data
# The original data was used to generate a hash as follows:
# sha256sum sampleList reference/*.fasta samples/*/*.fastq.gz > sha256sumCheck
# The command below checks the downloaded data (and the reference sequence) against the
# hashes that are saved in the sha256sumCheck file using sha256sum command, which is
# generally available on unix systems.
sha256sum -c sha256sumCheck
Step 2 - Prep work:
# Create files of sample directories and fastQ files:
ls -d samples/* > sampleDirectories.txt
rm sampleFullPathNames.txt 2>/dev/null
cat sampleDirectories.txt | while read dir; do echo $dir/*.fastq* >> sampleFullPathNames.txt; done
# Determine the number of CPU cores in your computer
numCores=$(grep -c ^processor /proc/cpuinfo 2>/dev/null || sysctl -n hw.ncpu)
Step 3 - Prep the reference:
cfsan_snp_pipeline index_ref reference/NC_011149.fasta
Step 4 - Align the samples to the reference:
# Align each sample, one at a time, using all CPU cores
export Bowtie2Align_ExtraParams="--reorder -X 1000"
export SamtoolsSamFilter_ExtraParams="-F 4 -q 30"
cat sampleFullPathNames.txt | xargs -n 2 -L 1 cfsan_snp_pipeline map_reads --threads $numCores reference/NC_011149.fasta
Step 5 - Find the sites having high-confidence SNPs:
# Process the samples in parallel using all CPU cores
export SamtoolsMpileup_ExtraParams="-q 0 -Q 13 -A"
export VarscanMpileup2snp_ExtraParams="--min-var-freq 0.90"
cat sampleDirectories.txt | xargs -n 1 -P $numCores cfsan_snp_pipeline call_sites reference/NC_011149.fasta
Step 6 - Identify regions with abnormal SNP density and remove SNPs in these regions:
cfsan_snp_pipeline filter_regions --window_size 1000 125 15 --max_snp 3 2 1 -n var.flt.vcf sampleDirectories.txt reference/NC_011149.fasta
Step 7 - Combine the SNP positions across all samples into the SNP list file:
cfsan_snp_pipeline merge_sites -n var.flt.vcf -o snplist.txt sampleDirectories.txt sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline merge_sites -n var.flt_preserved.vcf -o snplist_preserved.txt sampleDirectories.txt sampleDirectories.txt.PresVCF.filtered
Step 8 - Call the consensus base at SNP positions for each sample:
# Process the samples in parallel using all CPU cores
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline call_consensus -l snplist.txt --minConsDpth 3 --vcfFileName consensus.vcf --vcfRefName NC_011149.fasta -o XX/consensus.fasta XX/reads.all.pileup
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline call_consensus -l snplist_preserved.txt --minConsDpth 3 --vcfFileName consensus_preserved.vcf --vcfRefName NC_011149.fasta -o XX/consensus_preserved.fasta -e XX/var.flt_removed.vcf XX/reads.all.pileup
Step 9 - Create the SNP matrix:
cfsan_snp_pipeline snp_matrix -c consensus.fasta -o snpma.fasta sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline snp_matrix -c consensus_preserved.fasta -o snpma_preserved.fasta sampleDirectories.txt.PresVCF.filtered
Step 10 - Create the reference base sequence:
cfsan_snp_pipeline snp_reference -l snplist.txt -o referenceSNP.fasta reference/NC_011149.fasta
cfsan_snp_pipeline snp_reference -l snplist_preserved.txt -o referenceSNP_preserved.fasta reference/NC_011149.fasta
Step 11 - Collect metrics for each sample:
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline collect_metrics -o XX/metrics XX reference/NC_011149.fasta
Step 12 - Tabulate the metrics for all samples:
cfsan_snp_pipeline combine_metrics -n metrics -o metrics.tsv sampleDirectories.txt
Step 13 - Merge the VCF files for all samples into a multi-sample VCF file:
cfsan_snp_pipeline merge_vcfs -n consensus.vcf -o snpma.vcf sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline merge_vcfs -n consensus_preserved.vcf -o snpma_preserved.vcf sampleDirectories.txt.PresVCF.filtered
Step 14 - Compute the SNP distances between samples:
cfsan_snp_pipeline distance -p snp_distance_pairwise.tsv -m snp_distance_matrix.tsv snpma.fasta
cfsan_snp_pipeline distance -p snp_distance_pairwise_preserved.tsv -m snp_distance_matrix_preserved.tsv snpma_preserved.fasta
Step 15 - View and verify the results:
Upon successful completion of the pipeline, the snplist.txt file should have 2620 entries. The SNP Matrix can be found in snpma.fasta. The corresponding reference bases are in the referenceSNP.fasta file:
# Verify the result files were created
ls -l snplist.txt
ls -l snpma.fasta
ls -l snpma.vcf
ls -l referenceSNP.fasta
ls -l snp_distance_matrix.tsv
ls -l snplist_preserved.txt
ls -l snpma_preserved.fasta
ls -l snpma_preserved.vcf
ls -l referenceSNP_preserved.fasta
ls -l snp_distance_matrix_preserved.tsv
# Verify correct results
cfsan_snp_pipeline data agonaExpectedResults expectedResults
diff -q -s snplist.txt expectedResults/snplist.txt
diff -q -s snpma.fasta expectedResults/snpma.fasta
diff -q -s referenceSNP.fasta expectedResults/referenceSNP.fasta
diff -q -s snp_distance_matrix.tsv expectedResults/snp_distance_matrix.tsv
diff -q -s snplist_preserved.txt expectedResults/snplist_preserved.txt
diff -q -s snpma_preserved.fasta expectedResults/snpma_preserved.fasta
diff -q -s referenceSNP_preserved.fasta expectedResults/referenceSNP_preserved.fasta
diff -q -s snp_distance_matrix_preserved.tsv expectedResults/snp_distance_matrix_preserved.tsv
# View the per-sample metrics
xdg-open metrics.tsv
Step-by-Step Workflow - General Case¶
Note: the step-by-step workflows are not recommended. Most users should run the pipeline
with the all-in-one cfsan_snp_pipeline run command.
Step 1 - Gather data:
You will need the following data:
- Reference genome
- Fastq input files for multiple samples
Organize the data into separate directories for each sample as well as the reference. One possible directory layout is shown below. Note the mix of paired and unpaired samples:
./myProject/reference/my_reference.fasta
./myProject/samples/sample1/sampleA.fastq
./myProject/samples/sample2/sampleB.fastq
./myProject/samples/sample3/sampleC_1.fastq
./myProject/samples/sample3/sampleC_2.fastq
./myProject/samples/sample4/sampleD_1.fastq
./myProject/samples/sample4/sampleD_2.fastq
Step 2 - Prep work:
# Optional step: Copy your input data to a safe place:
cp -r myProject myProjectClean
# The SNP pipeline will generate additional files into the reference and sample directories
cd myProject
# Create file of sample directories:
ls -d samples/* > sampleDirectories.txt
# get the *.fastq or *.fq files in each sample directory, possibly compresessed, on one line per sample, ready to feed to bowtie
TMPFILE1=$(mktemp tmp.fastqs.XXXXXXXX)
cat sampleDirectories.txt | while read dir; do echo $dir/*.fastq* >> $TMPFILE1; echo $dir/*.fq* >> $TMPFILE1; done
grep -v '*.fq*' $TMPFILE1 | grep -v '*.fastq*' > sampleFullPathNames.txt
rm $TMPFILE1
# Determine the number of CPU cores in your computer
numCores=$(grep -c ^processor /proc/cpuinfo 2>/dev/null || sysctl -n hw.ncpu)
Step 3 - Prep the reference:
cfsan_snp_pipeline index_ref reference/my_reference.fasta
Step 4 - Align the samples to the reference:
# Align each sample, one at a time, using all CPU cores
export Bowtie2Align_ExtraParams="--reorder -X 1000"
export SamtoolsSamFilter_ExtraParams="-F 4 -q 30"
cat sampleFullPathNames.txt | xargs -n 2 -L 1 cfsan_snp_pipeline map_reads --threads $numCores reference/my_reference.fasta
Step 5 - Find the sites having high-confidence SNPs:
# Process the samples in parallel using all CPU cores
export SamtoolsMpileup_ExtraParams="-q 0 -Q 13 -A"
export VarscanMpileup2snp_ExtraParams="--min-var-freq 0.90"
cat sampleDirectories.txt | xargs -n 1 -P $numCores cfsan_snp_pipeline call_sites reference/my_reference.fasta
Step 6 - Identify regions with abnormal SNP density and remove SNPs in these regions:
cfsan_snp_pipeline filter_regions --window_size 1000 125 15 --max_snp 3 2 1 -n var.flt.vcf sampleDirectories.txt reference/my_reference.fasta
Step 7 - Combine the SNP positions across all samples into the SNP list file:
cfsan_snp_pipeline merge_sites -n var.flt.vcf -o snplist.txt sampleDirectories.txt sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline merge_sites -n var.flt_preserved.vcf -o snplist_preserved.txt sampleDirectories.txt sampleDirectories.txt.PresVCF.filtered
Step 8 - Call the consensus base at SNP positions for each sample:
# Process the samples in parallel using all CPU cores
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline call_consensus -l snplist.txt --minConsDpth 3 --vcfFileName consensus.vcf -o XX/consensus.fasta XX/reads.all.pileup
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline call_consensus -l snplist_preserved.txt --minConsDpth 3 --vcfFileName consensus_preserved.vcf -o XX/consensus_preserved.fasta -e XX/var.flt_removed.vcf XX/reads.all.pileup
Step 9 - Create the SNP matrix:
cfsan_snp_pipeline snp_matrix -c consensus.fasta -o snpma.fasta sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline snp_matrix -c consensus_preserved.fasta -o snpma_preserved.fasta sampleDirectories.txt.PresVCF.filtered
Step 10 - Create the reference base sequence:
# Note the .fasta file extension
cfsan_snp_pipeline snp_reference -l snplist.txt -o referenceSNP.fasta reference/my_reference.fasta
cfsan_snp_pipeline snp_reference -l snplist_preserved.txt -o referenceSNP_preserved.fasta reference/my_reference.fasta
Step 11 - Collect metrics for each sample:
cat sampleDirectories.txt | xargs -n 1 -P $numCores -I XX cfsan_snp_pipeline collect_metrics -o XX/metrics XX reference/my_reference.fasta
Step 12 - Tabulate the metrics for all samples:
cfsan_snp_pipeline combine_metrics -n metrics -o metrics.tsv sampleDirectories.txt
Step 13 - Merge the VCF files for all samples into a multi-sample VCF file:
cfsan_snp_pipeline merge_vcfs -n consensus.vcf -o snpma.vcf sampleDirectories.txt.OrigVCF.filtered
cfsan_snp_pipeline merge_vcfs -n consensus_preserved.vcf -o snpma_preserved.vcf sampleDirectories.txt.PresVCF.filtered
Step 14 - Compute the SNP distances between samples:
cfsan_snp_pipeline distance -p snp_distance_pairwise.tsv -m snp_distance_matrix.tsv snpma.fasta
cfsan_snp_pipeline distance -p snp_distance_pairwise_preserved.tsv -m snp_distance_matrix_preserved.tsv snpma.fasta
Step 15 - View the results:
Upon successful completion of the pipeline, the snplist.txt identifies the SNP positions in all samples. The SNP Matrix can be found in snpma.fasta. The corresponding reference bases are in the referenceSNP.fasta file:
ls -l snplist.txt
ls -l snpma.fasta
ls -l snpma.vcf
ls -l referenceSNP.fasta
ls -l snp_distance_matrix.tsv
ls -l snplist_preserved.txt
ls -l snpma_preserved.fasta
ls -l snpma_preserved.vcf
ls -l referenceSNP_preserved.fasta
ls -l snp_distance_matrix_preserved.tsv
# View the per-sample metrics
xdg-open metrics.tsv
Duplicate Read Removal¶
Prior to creating the pileup and calling snps, the pipeline detects and removes duplicate reads from the sample BAM files. When duplicates are found, the highest quality read among the duplicates is retained. Removing duplicate reads slightly reduces the depth of coverage in pileups and will sometimes impact the number of called snps. The number of called snps could either increase or decrease depending on whether reference-supporting or variant-supporting reads are removed. Removing duplicate reads impacts the subsequent application of virtually all snp filters: depth, variant allele frequency, strand bias, strand depth, and high density snp filtering.
Duplicate reads are removed with the Picard software tool which must be installed for this functionality.
You can disable this step and keep the duplicate reads by configuring the
RemoveDuplicateReads parameter in the configuration file.
You can customize the picard MarkDuplicates behavior to some extent by configuring the
PicardMarkDuplicates_ExtraParams parameter in the configuration file.
Duplicate read removal works best when the read names in the fastq files are in the original Illumina format.
When downloading fastq files from NCBI with fastq-dump, you should use the --origfmt command line option.
See Why are there no optical duplicates?
If you see No space left on device errors, you should set either the TMPDIR or TMP_DIR environment
variable to a directory with plenty of space for temp files:
export TMPDIR=/scratch/tmp
More information about the Picard MarkDuplicates tool can be found here:
- https://broadinstitute.github.io/picard/command-line-overview.html#MarkDuplicates
- http://gatkforums.broadinstitute.org/gatk/discussion/6747/how-to-mark-duplicates-with-markduplicates-or-markduplicateswithmatecigar
- http://broadinstitute.github.io/picard/faq.html
See also Configuration.
Local Realignment¶
When reads are mapped to the reference, each read is mapped independently. The reads may be misaligned around insertions or deletions. The local realignment process attempts to minimize the total number of mismatched bases in all the reads around the indels.
The reads are realigned with the GATK software (prior to version 4.0) which must be installed for this
functionality. It is a two step process. First, the pipeline identifies regions where indels are likely.
Then, the reads are realigned in the identified regions.
Local realignment can be a time-consuming process. You can disable this step by configuring
the EnableLocalRealignment parameter in the configuration file.
More information about the GATK indel realigner can be found here:
- https://software.broadinstitute.org/gatk/documentation/tooldocs/3.8-0/org_broadinstitute_gatk_tools_walkers_indels_IndelRealigner.php
- https://software.broadinstitute.org/gatk/documentation/article.php?id=38
See also Configuration.
SNP Filtering¶
The SNP Pipeline removes abnormal SNPs from the ends of contigs and from regions where many SNPs are found in
close proximity. The pipeline runs both ways, with SNP filtering, and without SNP filtering, generating
pairs of output files. You can compare the output files to determine which positions were filtered. The filtered output
files are named with the _preserved suffix, for example:
- snplist.txt : contains the unfiltered SNP positions with abnormal SNPs included
- snplist_preserved.txt : contains the filtered SNP positions without abnormal SNPs
- snpma.fasta : contains the unfiltered SNP matrix with abnormal SNPs included
- snpma_preserved.fasta : contains the filtered SNP matrix without abnormal SNPs
Other output files are named similarly.
The SNP filtering is performed by the filter_regions command. It runs after the phase 1 SNP detection and impacts
all subsequent processing steps.
Dense regions found in any sample are filtered from all the other samples by default. In this mode, if you add or remove
a sample from your analysis it may affect the final SNPs detected in all other samples. If you choose to do so, the dense
regions can be filtered from each sample individually with the --mode each command line option.
See FilterRegions_ExtraParams.
The drawing below depicts the behavior of the SNP Filtering.
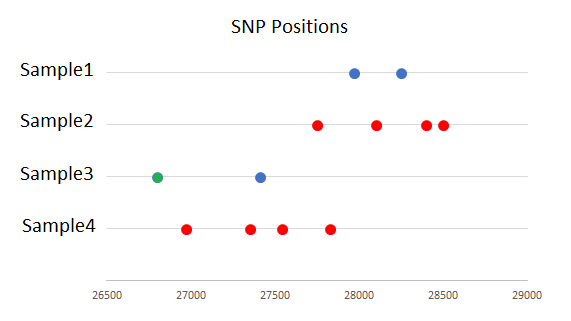
Sample 2 has enough snps in close proximity to form a dense region shown in red. These snps are filtered regardless of the
--mode option.
Sample 4 has enough snps in close proximity to form a dense region shown in red. These snps are filtered regardless of the
--mode option.
Sample 1 has two snps which by themselves do not form a dense region, but when the --mode all option is used, the snps
are engulfed by the dense region in sample 2 and removed.
Sample 3 has two snps which by themselves do not form a dense region. When the --mode all option is used, the blue snp
is engulfed by the dense region in sample 4 and removed. The green snp is not filtered regardless of the --mode
option because it is not within a dense region of any other sample. Merely being nearby a dense region in another
sample does not cause snp filtering.
The sensitivity of the SNP filtering can be controlled with parameters in the configuration file by setting values in
FilterRegions_ExtraParams. You can control the length of end-of-contig trimming, dense region window size, and
maximum snps allowed within the window. It’s possible to configure the SNP filter to find dense regions in multiple
window sizes, each with a different maximum allowed number of SNPs. For example, you can allow no more than 3 SNPs
per 1000 bases and 2 SNPs per 100 bases. See Configuration.
SNP Filtering With Outgroups¶
If there is an outgroup among the samples, you should configure the pipeline to exclude the outgroup samples from snp filtering. To exclude the outgroup samples:
First, make a file containing the sample ids of the outgroup samples, one sample id per line. The sample id is the name of the last subdirectory in the path to the sample:
SRR1556289
SRR1556294
Grab the default configuration file:
cfsan_snp_pipeline data configurationFile
Edit snppipeline.conf, and change the FilterRegions_ExtraParams parameter:
Add the --out_group option with the path to the file containing the outgroup sample ids.
Then run the snp pipeline with the -c command line options:
cfsan_snp_pipeline run -c snppipeline.conf -s mySamplesDir myReference.fasta
See also Configuration.
Excessive SNPs¶
Samples having many SNPs relative to the reference can slow the performance of the SNP Pipeline and greatly increase the size of the SNP matrix. The SNP Pipeline has the capability to exclude samples from processing when those samples have too many SNPs. This function excludes entire samples, not just regions within a sample. The samples with excessive SNPs exceeding a user-specified limit are excluded from the snp list, snp matrix, and snpma.vcf files.
There is also an indicator in the metrics file to identify the samples that have too many SNPs. A column in the
metrics.tsv file, Excluded_Sample, indicates when a sample has been excluded from the snp matrix. This column
is normally blank. See Metrics.
To exclude samples with excessive SNPs:
Grab the default configuration file:
cfsan_snp_pipeline data configurationFile
Edit snppipeline.conf, and change this setting:
MaxSnps=1000 # substitute your threshold value here, or -1 to disable this function
Then run the pipeline with the -c command line option:
cfsan_snp_pipeline run -c snppipeline.conf -s mySamplesDir myReference.fasta
See also Configuration.
Metrics¶
After creating the SNP matrix, the pipeline collects and tabulates metrics for all of the samples. The metrics are first collected in one file per sample in the sample directories. A subsequent step combines the metrics for all the samples together into a single tab-separated file with one row per sample and one column per metric. The tabulated metrics file is named metrics.tsv by default.
The metrics are:
| Metric | Description |
|---|---|
| Sample | The name of the directory containing the sample fastq files. |
| Fastq Files | Comma separated list of fastq file names in the sample directory. |
| Fastq File Size | The sum of the sizes of the fastq files. This will be the compressed size if the files are compressed. |
| Machine | The sequencing instrument ID extracted from the compressed fastq.gz file header. If the fastq files are not compressed, the machine ID is not captured. |
| Flowcell | The flowcell used during the sequencing run, extracted from the compressed fastq.gz file header. If the fastq files are not compressed, the flowcell is not captured. |
| Number of Reads | The number of reads in the SAM file. When using paired fastq files, this number will be twice the number of reads reported by bowtie. |
| Duplicate Reads | The number of reads marked as duplicates. These reads are not included in the pileup and are not used to call snps. When a set of duplicate reads is found, only the highest-quality read in the set is retained. |
| Percent of Reads Mapped | The percentage of reference-aligned reads in the SAM file. |
| Percent Proper Pair | The percentage of all reads in the SAM file that are aligned to the reference in the proper orientation and within the expected paired-end distance. The Percent Proper Pair metric is less than 100% when there are discordant alignments, unpaired alignments, or reads that are not mapped at all. This metric reflects the state of the alignment immediately after the mapping, however, subsequent steps of the pipeline remove the reads with low mapping quality, which has the effect of increasing the percentage of proper pairs prior to calling snps. |
| Average Insert Size | The average insert size of mapped paired reads in the SAM file as reported by SAMtools stats. Discordant alignments will increase the average insert size because by definition, discordant alignments are not within the expected paired-end distance. |
| Average Pileup Depth | The average depth of coverage in the sample pileup file. This is calculated as the sum of the depth of the pileup across all pileup positions divided by the number of positions in the reference. |
| Phase1 SNPs | The number of phase 1 SNPs found for this sample. The count is computed as the number of SNP records in the VCF file generated by the phase 1 snp caller (VarScan). |
| Phase1 Preserved SNPs | The number of phase 1 SNPs found by VarScan and preserved by
SNP Filtering. The count is computed as the number of SNP
records in the preserved VCF file generated by the
filter_regions command. |
| Phase2 SNPs | The number of phase 2 SNPs found for this sample. The count is computed as the number of SNP records in the VCF file generated by the consensus caller. |
| Phase2 Preserved SNPs | The number of phase 2 SNPs found for this sample and preserved by SNP Filtering. The count is computed as the number of SNP records in the preserved VCF file generated by the consensus caller. |
| Missing SNP Matrix Positions | The number of positions in the SNP matrix for which a
consensus base could not be called for this sample. The
inability to call a consensus base is caused by either a
pileup file with no coverage at a SNP position, or by
insufficient agreement among the pileup bases at the SNP
position. The minimum fraction of reads that must agree at a
position to make a consensus call is controlled by the
minConsFreq parameter. |
| Missing Preserved SNP Matrix Positions | The number of positions in the preserved SNP matrix for which
a consensus base could not be called for this sample. The
inability to call a consensus base is caused by either a
pileup file with no coverage at a SNP position, or by
insufficient agreement among the pileup bases at the SNP
position. The minimum fraction of reads that must agree at a
position to make a consensus call is controlled by the
minConsFreq parameter. |
| Excluded Sample | When a sample has an excessive number of snps exceeding the
MaxSnps parameter value, this metric will have the value
Excluded. Otherwise, this metric is blank. |
| Excluded Preserved Sample | When a sample has an excessive number of preserved snps
exceeding the MaxSnps parameter value, this metric will
have the value Excluded. Otherwise, this metric is blank. |
| Warnings and Errors | A list of warnings or errors encountered while collecting the metrics. |
Error Handling¶
The SNP Pipeline detects errors during execution and prevents execution of subsequent
steps when earlier steps fail. A summary of errors is written to the error.log file.
Detailed error messages are found in the log files for each process.
See Logging.
By default, the SNP Pipeline is configured to stop when execution errors occur. However, it is
possible some errors may affect only individual samples and other samples can still be
processed. If you want the pipeline to continue processing after an error affecting only
a single sample has occurred, you can try disabling the StopOnSampleError
configuration parameter (not recommended). See Configuration.
When StopOnSampleError is false
the pipeline will attempt to continue subsequent processing steps when an error does not
affect all samples. Errors are logged in the error.log file regardless of how the
StopOnSampleError parameter is configured. You should review the error.log
after running the pipeline to see a summary of any errors detected during execution.
Note: currently, when using the Torque job queue manager, the pipeline will always stop on
errors regardless of the StopOnSampleError parameter setting.
When errors stop the execution of the pipeline on a High Performance Computing cluster, other non-failing jobs
in progress will continue until complete. However, subsequent job steps will not execute and
instead will remain in the queue. On Grid Engine, the qstat command will show output like
the following:
3038927 0.55167 mapReads app_sdavis Eqw 07/15/2017 16:50:03
3038928 0.00000 callSites app_sdavis hqw 07/15/2017 16:50:04
3038929 0.00000 filterRegi app_sdavis hqw 07/15/2017 16:50:04
3038930 0.00000 mergeSites app_sdavis hqw 07/15/2017 16:50:04
3038931 0.00000 callConsen app_sdavis hqw 07/15/2017 16:50:04
3038932 0.00000 snpMatrix app_sdavis hqw 07/15/2017 16:50:04
3038933 0.00000 snpReferen app_sdavis hqw 07/15/2017 16:50:04
3038934 0.00000 mergeVcfs app_sdavis hqw 07/15/2017 16:50:05
3038935 0.00000 distance app_sdavis hqw 07/15/2017 16:50:05
3038936 0.00000 collectMet app_sdavis hqw 07/15/2017 16:50:05
3038937 0.00000 combineMet app_sdavis hqw 07/15/2017 16:50:05
To clear the jobs from the queue on Grid Engine:
seq 3038927 3038937 | xargs qdel